
Pipeline News and Updates

|
A very busy week, with a new flu drug approval and lots research announced at all medical meetings.
In time for the new flu season the FDA approved baloxavir marboxil (Xofluza from Roche and Shionogi) on 10/24/18 for the treatment of acute uncomplicated influenza in patients 12 year or older that have been symptomatic for 48 hours or less. Data from 55 patients in 3 Phase I & II trials involving 17 unique TRK fusion–positive tumor types, suggesting that after 1-year 71% of patients treated with larotrectinib had an ongoing response and 55% remained progression-free. Loxo announced that after two years follow-up, the 55 patients had an overall response rate (ORR) of 80%, partial response rate (PRR) of 62% and a complete response rate (CRR) of 18%. In a group of 67 TRK fusion patients, the ORR was 81%, PFF of 65% and CRR of 17%. When the results of both the 55 patient and 67 patient groups were analyzed together (122 patients), the ORR was 81%, PFF of 63% and CRR of 17%. In a 24 pediatric patient, Phase I trial, larotrectinib was well tolerated and almost all patients with TRK fusion positive cancers had an objective response. ImmunoGen announced that in the 56 patient, Phase Ib/II, FORWARD II trial, the overall response rate was 30% and progression-free survival was 4.2 months in patients with platinum-resistant epithelial ovarian cancer treated with mirvetuximab soravtansine plus pembrolizumab. CHMP recommended approval of Sanofi’s Dengue vaccine AbbVie announced that in the 26-week, 1,629 patient, Phase III, SELECT-COMPARE trial, more patients treated with upadacitinib/methotrexate improved compared to adalimumab/methotrexate or methotrexate monotherapy achieving ACR20 (67%, 57%, 36%), ACR50 (54%, 42%, 21%) and ACR70 (35%, 23%, 10%) in rheumatoid arthritis patients. Novartis announced that in the 94 patient, open label, Phase II GEOMETRY trial, treatment with capmatinib resulted in an overall response rate of 72% in treatment-naive patients and 39% in previously treated patients with advanced MET exon-14 skipping mutated non-small cell lung cancer. DBV Technologies submitted a BLA for Viaskin Peanut in October 2018. Pfizer and Lilly announced that a 16-week, 698 patient, Phase III trial, where treatment with tanezumab 5 mg resulted in a 57% of patients having a 50% improvement in the WOMAC score compared to a 50% with 2.5 mg and a 38% with placebo in patients with osteoarthritis pain of the knee and hip. Rapidly progressive osteoarthritis (RPOA) was observed in 1.3% of tanezumab compared to none with placebo. Most of the RPOA was Type 1, which is characterized by joint space narrowing as compared to the more serious type 2 that is more rapid and leads more often to bone loss and joint destruction. Pfizer and Lilly have 5 ongoing Phase III studies evaluating tanezumab in the treatment of in OA, chronic lower back pain and cancer pain. Results are expected in the first half of 2019. J&J announced announced preliminary data from the 198 patient, Phase I/IIa TRAVERSE trial suggested that a tetravalent version of the HIV vaccine produced a broadener the immunologic response compared to the trivalent vaccine. Based on preliminary results from the TRAVERSE study demonstrating a broader immunologic response compared the the trivalent vaccine, J&J will advance the tetravalent vaccine into a 2,600 patient Phase IIb trial. BeyondSpring announced that in a 72 patient, Phase II trial, 50% of patients that received plinabulin and pegfilgrastim experienced neutropenia compared to 81% that received pegfilgrastim monotherapy in patients treated with docetaxel, doxorubicin and cyclophosphamide. In the 110 patients, Phase II ARADES trial, darolutamide elicited a PSA responses in about a third of men with progressive metastatic castration-resistant prostate cancer. Findings demonstrated that darolutamide monotherapy in this particular group of men provided disease suppression. Bayer and Orion announced preliminary results from the 1,500 patient, Phase II ARAMIS trial, where treatment with darolutamide improved metastasis-free survival compared to placebo in men with non-metastatic castration-resistant prostate cancer. Biogen announced that in an 18-month, 856 patient, Phase II trial, BAN2401 slowed cognitive decline and reduced beta-amyloid deposits in patients that received the highest dose. The drug had failed a Bayesian statistical analysis at 12 months, but at 18 months, a conventional statistical analysis found a benefit at 6, 12 and 18 months. The findings were criticized because the highest dose group had few patients with a APOE4 mutation that increases the risk for developing Alzheimer’s. Biogen announced that a subgroup analysis suggested that patients with the mutation did better with BAN2401 than those without. However, only 10 patients in the high dose group had the mutation compared to 113 patients with the mutation that received placebo. Scynexis has two Phase III trials evaluating oral ibrexafungerp for the treatment vulvovaginal candidiasis (VVC) and one trials evaluating the drug for prophylaxis of VVC. Mild to moderate thrombotic events were identified in a Phase I trial of the injectable form of the drug. Lundbeck announced preliminary information from the 10-week, 697 patient, Phase III, DAYBREAK trial Lu AF35700 was similar to olanzapine and risperidone in patients with treatment-resistant schizophrenia, which missed the primary endpoint of superiority vs conventional therapy. Ultragenyx announced that in a 44 patient, Phase III trial, triheptanoin was no better than placebo in reducing paroxysmal movements in patients with glucose transporter type 1 deficiency syndrome. After the drug failed the Phase III trial, Ultragenyx has stopped development of triheptanoin as a therapy for glucose transporter type-1 deficiency syndrome (Glut1 DS), but will continue to develop the drug for long-chain fatty acid oxidation disorders. In the 8-week, 109 patient, Phase II, DUET trial of sparsentan in focal segmental glomerulosclerosis patients, sparsentan reduced proteinuria by 44.8% compared to 18.5% with irbesartan in patients with focal segmental glomerulosclerosis. In a 42 patient, Phase II trial, treatment with selinexor in combination with low-dose bortezomib and dexamethasone resulted in a 63% overall response rate and progression-free survival of 9 months in patients with relapsed or refractory multiple myeloma. In a 12 week, Phase I trial, AADvac1 induced an immunological response to pathological tau proteins. No safety issue occurred during the trial and cognition remained stable. In the 72-week, 26 patient, open label FUNDAMANT extension trial, responders retained an immunoglobulin G (IgG) antibody response against the tau peptide at 6-months, but booster doses were required to restore IgG levels. Five patients withdrew from the trial, but there was a trend of slower atrophy in MRI evaluation and lower decline in cognitive assessment in patients with high titers. In the 12-week,116 patient Phase II, TORTUGA trial, patients treated with filgotinib had a 1.5 point decrease in their Ankylosing Spondylitis Disease Activity Score (ASDAS) compared to a 0.6 decrease with placebo. A 24-week, 447 patient trial that studied the use of teneligliptan as an add on to metformin therapy showed a 0.3% to 0.63% reduction in HbA1C compared to placebo. In a 12-week, 324 diabetic patient trial, teneligliptan reduced HbA1C by 0.9% to 1% more than placebo. In a 12-week, 194 patient, Phase III trial teneligliptan lowered HbA1C 1% more than placebo. Mitsubishi Tanabe announced that in a 24-week, 447 patient trial that studied the use of teneligliptan as an add on to metformin therapy, teneligliptan demonstrated a 0.3% to 0.63% reduction in HbA1C compared to placebo. Mitsubishi Tanabe announced that in a 12-week, 324 diabetic patient trial, teneligliptan reduced HbA1C by 0.9% to 1% more than placebo. Mitsubishi Tanabe announced that in a 12-week, 194 patient, Phase III trial teneligliptan lowered HbA1C 1% more than placebo. In a 12-week, 201 patient, Phase III trial, teneligliptan had a similar reduction in HbA1C 1% compared to sitagliptin (-1.02 vs -1.03) when added to metformin and glimepiride in patients with type 2 diabetes (T2DM) inadequately controlled with metformin and glimepiride. In a 7-week, 126 patient trial, treatment with cebranopadol resulted in 2.81 mg of daily rescue morphine immediate release given daily over the last 2 weeks of treatment versus morphine in patients with moderate to severe cancer-related pain. Novo announced that in the 52-week, 731 patient, Phase III Pioneer-8 trial, treated with oral semaglutide 7mg and 14mg lowered HbA1c 0.5 to 1.2% compared to 0% with placebo and weight loss of 1 to 4.3 Kg compared to a 0.6mg gain with placebo in patients with type 2 diabetics. In a 452 patient, Phase II trial, cefiderocol was non-inferior to imipenem/cilastatin in clinical cure and microbiological eradication in hospitalized adult UTI patients. Cefiderocol has demonstrared activity againset highly resistant Gram-negative bacteria, but has not activity against Gram Positive bacteria. Eisai announced that in the 4-week, 62 patient, Phase III trial, lemborexant improved 24-hour circadian rhythm pattern reduced nighttime activity compared to placebo in patients with irregular Sleep-Wake Rhythm Disorder (ISWRD) in patients with mild to moderate Alzheimer’s disease. Stay current with drugs in the late stages of development with the Prescribe Right Pharmaceutical Pipeline Tracker. There are three types of opioid receptors (mu, kappa, delta). Activation of the opioid receptors stimulate G proteins which initiate the intracellular communication process leading to the physiologic responses.
All three opioid receptors cause analgesia and respiratory depression. Kappa and Mu receptors also cause sedation. Mu receptors can be divided into two types, Mu1 and Mu2. Stimulation of Mu1 receptors cause analgesia, bradycardia and respiratory depression, while Mu2 receptors cause respiratory depression, physical dependence and euphoria. Morphine and synthetic opioids are mu-opioid receptor agonists. Three selective Mu-opioid receptor agonists are in the late stages of development with two having PDUFA dates through May 2019. None of the three have FDA Priority Designations. Oliceridine (Olinvo fromTrevena) PDUFA: November 2, 2018 Oliceridine is being developed by Trevena. An FDA review of oliceridine felt that data did not support the drug having less respiratory depression than morphine and the highest dose (0.5mg) appeared to be less efficacious than morphine in analgesic effects, but similar in ADR. The report also noted that lower doses of oliceridine had fewer ADR, but less analgesic effects. All strengths of olicerdine provided more analgesic effect than placebo. An early study provided evidence that dosing is not affected by renal impairment. Oliceridine will most likely be a schedule II drug. An FDA advisory committee voted 8-7 to not recommend approval of oliceridine and recommended additional clinical trials. The committee also noted the potential for QT prolongation. Trevena announced that top line results from two Phase III oliceridine trials found the drug to be superior to placebo in patients with moderate-to-severe acute pain. The 0.35 mg and 0.5 mg doses of oliceridine demonstrated efficacy comparable to morphine within 24-48 hours. Following a 1.5 mg loading dose, all oliceridine dose regimens (0.1 mg, 0.35 mg, and 0.5 mg) were able to display rapid onset with statistically significant efficacy by 5 to 15 minutes. Our knowledgebase contains no published studies about oliceridine. Sufentanil Sublingual (Dsuvia from AcelRx Pharmaceuticals) PDUFA: November 3, 2018 Dsuvia, being studied for being studied for post-op and battlefield pain, is a sublingual tablet administered by a pen like applicator. In a review the FDA found sublingual sufentanil tablets to be efficacious for moderate-to-severe acute pain but had concerns over the maximum dosing and the risk of misplaced tablets due to their small size. AcelRx and the FDA have both proposed a REMS program for sublingual sufentanil tablets. The FDA’s Anesthetic and Analgesic Drug Products Advisory Committee voted 10-3 to recommend approval of sublingual sufentanil tablets. In a 76 patient, Phase III trial, patients treated with sublingual sufentanil experienced a mean decrease in pain intensity of 35% at 1 hour. In a 143 patient, phase III trial, sublingual sufentil demonstrated efficacy compared to placebo in control of outpatient abdominal surgery pain. Our knowledgebase contains seven published studies for sublingual sufentanil. Click on this link for one-month’s access to the Pharmaceutical Pipeline Tracker @ $115 with no long-term commitment: https://www.prescriberight.com/subscription-offers.html then sign into www.prescriberight.com with your user name and password to conduct a single drug search for sufentanil to view the studies. NKTR-181 (Nektar)PDUFA: May 28, 2019 NKTR-181 being studied for moderate to severe chronic low back pain has reduced permeability across the blood-brain barrier, so it may less euphoria and sedation. Nektar announced that NKTR-181 reduced pain compared to placebo in 600 opioid-naive patients with moderate to severe chronic low back pain. A small trial suggested that NKTR-181 had a lower abuse potential than oxycodone. There is one published study in our knowledgebase which concluded that NKTR-181 demonstrated delayed onset of CNS effects and significantly lower abuse potential scores compared with oxycodone in recreational opioid users. We evaluate over 1,000 data feeds regarding investigational drugs to keep information for 630 drugs in our database current, so you can stay up to date. Here is a summary of the significant developments from last week.
The FDA approved Medivation’s talazoparib (Talzenna) on October 16, nearly a month ahead of its PDUFA Date, for the treatment of advanced BCRA-positive breast cancer. We list two published studies for Talazoparib. Try us for a month to review talazoparib’s published studies and learn about an additional twenty breast cancer investigational drugs in the Pharmaceutical Pipeline Tracker. Click on this link for one-month’s access to the Pharmaceutical Pipeline Tracker @ $115 with no long-term commitment: https://www.prescriberight.com/subscription-offers.html An FDA Advisory Committee voted 10-0 to recommend prucalopride tablets for the treatment of adults with chronic idiopathic constipation. In a review of Shire and Janssen’s prucalopride, the FDA noted that despite the drug being available in Europe since 2009, there have been no controlled trials lasting at least 12 months. In another FDA review, an integrated safety analysis of 2552 patients, the most common ADR in prucalopride trials were gastrointestinal disorders including nausea, diarrhea, abdominal pain, and headache with similar cardiovascular events compared to placebo. A final decision on prucalopride is expected in December. The drug does not have a PDUFA Date nor any Priority Designation. Alnylam is planning to begin a rolling NDA for givosiran by the end of the year with completion in mid-2019, after full results from a Phase III trial are available. Givosiran has Orphan Drug and Breakthrough Therapy priority designations. MedDay announced that in a 12-month, 220 patient, open label trial, treatment with high dose biotin improved the Expanded Disability Status Scale (EDSS) and timed 25-foot walk (T25W) from baseline to one year in patients with progressive multiple sclerosis or secondary progressive multiple sclerosis. CHMP did not recommend approval of high dose biotin for MS due to a lack of clinical evidence. Sarepta licensed U.S. and non-European marketing rights for LYS-SAF302 from Lysogene. There is one citation in the Pharmaceutical Pipeline Tracker. There are twenty additional gene therapy investigational drugs in our Knowledgebase. Try us for a month to review Lysogene’s published finding and some of the other over 800 additional clinical studies in our Knowledgebase. Click on this link for one month’s access to the Pharmaceutical Pipeline tracker @ $115 with no long-term commitment: https://www.prescriberight.com/subscription-offers.html Search for gene therapy under the Indication Search button to see the list of twenty-one gene therapy investigational drugs and review each drug individually. Celgene announced that in a Phase III trial, treatment with ozanimod resulted in a lower relapse rate and fewer brain MRI lesions compared to interferon beta-1a in patients with relapsing multiple sclerosis. The company also announced that in a pooled analysis of the phase III trial and another 24-month trial, ozanimod reduced the annual relapse rates by 43% with a 1 mg dose and 26% with a 0.5 mg dose compared to interferon beta-1a in 2,666 patients with relapsing multiple sclerosis. Mylan and Theravance announced that in a 28-day, 207 patient, Phase III trial, nebulized revefenacin had similar improvements in trough FEV1 and trough FVC to MDI tiotropium in patients with COPD. The drug has a Nov 13, 2018, PDUFA Date. In a pooled analysis of three Phase II trials and a Phase I trial, nebulized revefenacin did not prolong QT interval nor increase risk of major adverse cardiac events (MACE). AZ announced that in a Phase III trial, the addition of selumetinib to radioactive iodine did not improve complete remission compared to radioactive iodine alone in patients with nonmetastatic differentiated thyroid cancer that were post total thyroidectomy. Selumetinib was granted an Orphan Drug Priority Designation by the FDA. Eisai announced that in a Phase III trial, lemborexant reduced sleep latency compared to placebo. In a Phase III trial the most common ADR with lemborexant were headache, somnolence and influenza. Allergan announced the most common ADR with ubrogepant in a 1,230-patient open label extension safety trial were nasopharyngitis, upper respiratory tract infection, sinusitis, urinary tract infection, and influenza. In a second safety trial, involving 516 patients, the most common ADR were headache, oropharyngeal pain, and nasopharyngitis. No hepatic safety concerns were noted in either safety trial. Allergan plans to submit an NDA for ubrogepant in 1Q19, for migraine prophylaxis. Four additional drugs of the eight migraine drugs still in development are being studied for migraine prophylaxis. The New England Journal of Medicine published two trials of cystic fibrosis transmembrane conductance regulator (CFTR) correctors being developed by Vertex. In a Phase II study, VX-445 in combination with tezacaftor/ivacaftor increased FEV1 by 8 to 14% compared to no improvement with placebo. In the second Phase II study, VX-659 in combination with tezacaftor/ivacaftor increased FEV1 by 10 to 13% compared to 0.4% improvement with placebo. In both Phase II trials for VX-445 or VX-659, ADR were mostly mild to moderate with no dose-limiting side effects or toxic effects. Researchers estimate the combination of either VX-445 or VX-659 in combination with tezacaftor/ivacaftor could treat the underlying cause of CFTR in 90% of cystic fibrosis patients. Vertex has three ongoing trials for each drug in combination with tezacaftor/ivacaftor for the treatment of cystic fibrosis. Novartis has the ongoing Phase III study evaluating intravenous AVXS-101 in spinal muscular atrophy Type 1, 2 & 3 and a Phase 1 study evaluating AVXS-101 in spinal muscular atrophy Type 2. AVXS-101 will compete with nusinersen. Novartis, which purchased AveXis in April, has submitted a BLA and MAA. AVXS-101 does not have a PDUFA Date but has orphan drug, breakthrough therapy and fast track priority designations. The other investigational drug for SMA is Catalyst/BioMarin Pharmaceutical’s amifampridine (Firdapse) with a PDUFA Date of Nov 28, 2018. Amifampridine has orphan drug and breakthrough therapy Priority Designations. Amifampridine is approved in the EU. There are nine published studies for amifampridine and one published study for AVXS-101 in our knowledgebase. Click on this link for one month’s access to the Pharmaceutical Pipeline tracker @ $115 with no long-term commitment to review both drugs’ published studies: https://www.prescriberight.com/subscription-offers.html Novartis announced that in a Phase III trial, alpelisib in combination with fulvestrant was not significantly better than fulvestrant monotherapy in the overall population of men and postmenopausal women with HR+/HER2- advanced breast cancer. In a subset of 341 who harbored PIK3CA mutations, PFS was 11 months with alpelisib and 5.7 months with placebo. The FDA found AcelRx Pharmaceuticals’ sublingual sufentanil tablets to be efficacious for moderate-to-severe acute pain but had concerns over the maximum dosing and the risk of misplaced tablets due to their small size. AcelRx and the FDA have both proposed a REMS program for sublingual sufentanil tablets. The FDA’s the Anesthetic and Analgesic Drug Products Advisory Committee voted 10-3 to recommend approval of sublingual sufentanil tablets.
The NDA and MAA have been accepted for AbbVie’s sponimod for the treatment of Type 2 Diabetes Mellitus, Nephropathy. The FDA is expected to make a decision on sponimod in March 2019. The FDA rejected Acacia Pharma’s amisulpride’s NDA on 10/8/18 due to manufacturing issues. An FDA review of Trevena’s oliceridine was skeptical that the drug had less respiratory depression than morphine. The highest dose (0.5mg) appeared to be less efficacious than morphine in analgesic effects, but similar in ADR. Lower doses had few ADR, but less analgesic effects. All strengths of oliceridine provided more analgesic effect than placebo. The FDA’s Anesthetic and Analgesic Drug Products Advisory Committee voted 8-7 against recommending approval of oliceridine ahead of the November 2, 2018 PDUFA Date. Ardelyx announced that in a Phase III, safety trial, the most common ADR with tenapanorwas diarrhea. In the 2-year, 837 patient, Phase III, VOYAGER 1 trial, patients were randomized to placebo, guselkumab or adalimumab. After 16 weeks, placebo patients were switched to guselkumab. After 52 weeks adalimumab patients were switched to guselkumab. At 100 weeks, results were similar among patients that switched from placebo or adalimumab to guselkumab. Efficacy was measured using the Psoriasis Area and Severity Index (PASI) and the Investigator's Global Assessment (IGA) scales. Overall PASI 75 was achieved by 94.8% of patients, PASI 90 by 82.1%, PASI 100 by 49.0%, IGA 0/1 by 82.4% and IGA 0 by 53.8% at week 100 in patients with moderate to severe psoriasis. In a 6-month Phase I/II trial, Pluristem Therapeutics’ PLX-PAD increased muscle volume by 300% and muscle force by 500% in patients with injured gluteal muscles after hip replacement. 2-year data from the study found better functional improvement with the lower dose most likely due to increased late‐onset immune reactivity with the higher dose. Menlo announced that in a Phase II trial, serlopitant did not improve 24-hour cough frequency compared to placebo in patients with chronic cough. J&J has an ongoing Phase III trial testing esketamine for treatment-resistant depression and major depressive disorder with imminent risk of suicide. J&J filed an NDA for esketamine in September 2018 and an MAA in October 2018. The drug does not have a PDUFA Date but does have a Breakthrough Therapy Priority Designation. Find more information about the efficacy and safety of esketamine along with links to two studies in PubMed in the Pharmaceutical Pipeline Tracker for as little as $115 per month with no annual commitment. https://www.prescriberight.com/subscription-offers.html Last spring, I looked at the trend in the number of new drug approvals by the FDA. The FDA was coming off a year with a greatly increased number of approvals compared to the previous year. When looked at over time, it appeared this was a long term trend that built on streamlining and optimizing the drug approval process. Thus, 2017 was similar to 2015 and just a little higher than 2012 and 2014. So, compared to 2016, 2017 looked more in line with the 2014-2015 trend in drug approvals and 2016 had a reduced number compared to previous years. 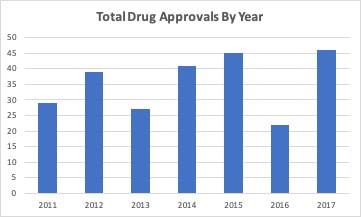 After one quarter, it appeared that 2018 was going to look more like 2016 with fewer drug approvals. I even wrote an editorial suggesting this but cautioned that it was still early to forecast the final volume. 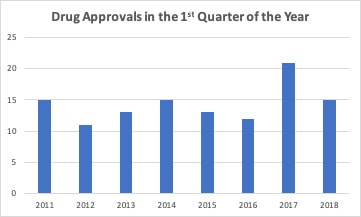 The FDA has been updating guidance that it issues for the development of drugs. As an example, new guidance for five neurological conditions was released at the beginning of the year. This new guidance along with new types of clinical trial methodologies such as adaptive trials have has effectively brought more drugs into the final stages of development. The FDA approved a greater number of drugs each month from May to September in 2018. The only exception was June, when they equaled the previous highest total. Through three quarters, the FDA is now on track to exceed the number of drugs approved in 2017 with the highest number of total approvals in seven years. 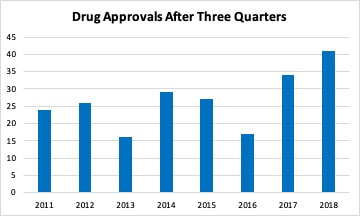 There has also been an increase in the number of generic drugs approved. The FDA uses an October to September fiscal year and reported 781 drugs approved in fiscal year 2018 compared to 763 in fiscal 2017. As with branded drugs, the FDA has also been releasing guidance documents to streamline approval of generic drugsand to promote competition for single source drugs with no patent protection. The FDA’s generic approval program should increase the number of manufacturers and help stabilize drug supplies and lower prices.
Subscribe to the Pharmaceutical Pipeline Tracker for only $115 per month to take a deeper dive into the investigational drug universe: https://www.prescriberight.com/subscription-offers.html Search for budget busting drugs by clicking on our “Current list of drugs with PDUFA”date button to review upcoming approval deadlines and any drugs with priority designations. Follow this link to view our case studies: https://www.prescriberight.com/case-studies.html Contact us with specific requests or questions at https://www.prescriberight.com/contact.html With the approval of fremanezumab and emgality in September, the FDA has approved three CGRP inhibitors. All three drugs are administered by subcutaneous (subQ) injection, but differ in their dosage forms and injection frequency.
* 140 mg is administered as 2 consecutive 70 mg subQ injections
** Given as 3 consecutive 225 mg subQ injections *** 240 mg is administered as 2 consecutive 70 mg subQ injections Erenumab and galcanezumab are administered monthly and available as either an auto-injector or prefilled syringe. Fremanezumab can be given monthly or quarterly. Because the drug is more viscous, it is currently only available as a prefilled syringe, but Teva is working on an auto-injector. All three drugs are only available in a single strength. A higher dose (erenumab and fremanezumab), therefore require multiple injections. All three drugs are well tolerated. The most common adverse effect with all three drugs is injection site reactions. Erenumab also lists constipation as a common adverse effect. The Wholesale Acquisition Cost (WAC) for one syringe is $575 for all three drugs ($1,725 per quarterly dose of femanezumab) for an annual WAC of $6,900. Each company is also offering a program to cover the copay for patients with commercial insurance. Express Scripts is pushing CGRP inhibitor manufacturers to set a lower price for their drug, rather than set a high price and offer a rebate. Express Scripts also wants to set up performance contract for the drugs based on efficacy on use of the drug in their patients. ICER released a final review of CGRP inhibitors on July 3, 2018. ICER found insufficient evidence to recommend erenumab or fremanezumab over oral preventative drugs or botulism toxin for prevention of migraine in untreated patients. ICER did find evidence of a benefit for use of erenumab or fremanezumab in patients that had previously failed preventative therapy for chronic migraine. Data was supportive but inconclusive for prevention of episodic migraine. ICER found insufficient evidence for use of galcabezumab in either indication. The drugs were found to be safe with the most common ADR of injection site reactions and upper respiratory symptoms. CGRP inhibitors were estimated to improve quality of life years (QALY) for episodic and chronic migraine patients. With an announced WAC price of $6,900/year for erenumab, ICER estimates an annual cost of $5,000/year after discounts. ICER estimates a price of $3,700 to $5,300 per year to be cost effective. Erenumab or fremanezumab were found to be cost effective in QALY gained in patients that had failed at least one preventative therapy. The estimated cost per QALY were lower for chronic migraine compared to episodic migraine. ICER felt that insurers would be justified in setting limits or restrictions on CGRP inhibitors due to insufficient long-term safety data and high cost. With pricing equivalent month the three drugs, we can expect to see more aggressive marketing and pricing as they battle for market share. There are currently no comparative trials between the three new drugs or comparing an CGRP-inhibitor to older oral drugs. Approvals
The FDA rejected ferric maltol during Shield’s first submission, but the company has resubmitted an NDA, which was accepted by the FDA this month. Tocagen announced that in a Phase I trial, patients treated with Toca 511 and Toca FC had an overall survival rate of 35% at 2 years and 26% at 3 years. Toca 511 and Toca FC have FDA Breakthrough Therapy Priority Designations. The EMA designated Toca 511 & Toca FC for orphan status. Viking announced top line results from a Phase II trial in patients recovering from hip fracture surgery. The highest dose of VK5211 (2 mg) increased lean body mass 9.1%, decreased fat mass 6.2% and increased 6-minute walking distance by 22 meters compared to placebo. At 24 weeks, increased lean body mass was maintained, but was similar to placebo. In a crossover trial, Jazz’s solriamfetol, PDUFA Date of Dec 20, 2018, was found to have abuse potential that was similar to or lower than phentermine. The drug has an Orphan Drug Priority Designation. ObsEva announced that in a Phase III trial, nolasiban improved the live birth rate 25% to 35% compared to placebo. ObsEva plans to file an MAA in the EU in 2019. Arena announced that in a Phase II trial in patients with pulmonary arterial hypertension (PAH), ralinepag demonstrated a 29.8% improvement in pulmonary vascular resistance compared to placebo. Arena announced interim data from their open label extension of their Phase II PAH trial, where treatment with ralinepag continued to demonstrate an improvement in pulmonary vascular resistance and 6-minute walk distance in both patients that continued on ralinepag (1.8 years) and patients that crossed over from placebo (1.4 years). The drug has an Orphan Drug Priority Designation. Current trial data display no significant safety concerns; no independent safety data are yet available. The most common ADR reported for ralinepag in a Phase II trial were headache and nausea. Omeros announced that in a Phase II trial, data suggested OMS721 improved kidney function and a reduced corticosteroid dose in IgAN, membranous nephropathy, lupus nephritis, and complement component 3 (C3) glomerulopathy. The drug has Fast Track, Breakthrough Therapy and Orphan Drug Priority Designations. Current trial data display no significant safety concerns; no independent safety data are yet available. The most common ADR reported for OMS721 in a Phase II trial were fatigue and anemia. Omeros has anongoing Phase III trial evaluating OMS721 in the treatment of Immunoglobulin A (IgA) Nephropathy and as a treatment for glomerulonephropathies and hematopoietic stem cell transplant-associated thrombotic microangiopathy. Roche announced that in a Phase III trial, a single dose of baloxavir marboxil reduced the time to alleviation of symptoms by 29.1 hours compared to placebo and was similar to 5 days of oseltamivir in influenza patients at high risk for complications. Baloxavir marboxil also reduced viral shedding more rapidly than oseltamivir or placebo. PDUFA Date: 12/24/2018. In the safety study that was part of the Phase III trial, nausea was less common with a single dose of baloxavir marboxil than 5 days of oseltamivir. Sarepta announced interim results from the first 4 patients in a 27 patient, Phase I/II trial, where microdystrophin gene therapy increased micro-dystrophin levels to 74% of normal dystrophin levels, though it is not currently known if microdystrophin has the same functional benefit as normal dystrophin. Ibudilast was granted orphan status by the FDA. The FDA granted a priority review for Karyopharm Therapeutics’ selinexor and assigned a PDUFA date of April 6, 2019. Stay current with drugs in the late stages of development with the Prescribe Right Pharmaceutical Pipeline Tracker. For the second month in a row the FDA pushed through five new drug approvals. This was accomplished by a surge of activity during the last week of the month for cancer and migraine drugs. The FDA rejected one drug, and another failed to produce its expected results. There were updates to nine drugs with PDUFA Dates and Priority Designations (potential budget busters). Read on for the details.
APPROVALS
31 citations were added to our knowledgebase bringing the total to 811. Each citation is accessible from the single drug look-up monograph via a URL link to the publication. There are now 612 investigational drugs in the Knowledgebase. We launched our new “use us when you need us” subscription plans. Click on this link for a preview. https://www.prescriberight.com/access-pharmaceutical-pipeline-tracker.html Here are three additional links to the Use Cases we’ve developed: https://www.prescriberight.com/monitoring-drugs-with-pdufa-dates.html https://www.prescriberight.com/monitoring-by-indication.html How to gather information about Right to Try requests: https://www.prescriberight.com/research-a-single-drug.html Four FDA approvals during the last week in September:
|
Stay informed, subscribe to the
Prescribe Right Pharmaceutical Pipeline Tracker Latest Tweets from Prescribe Right
Archives
July 2023
|
 RSS Feed
RSS Feed