
Vaccine manufacturer |
BioNTech/Pfizer |
Moderna |
Johnson & Johnson |
Astrazeneca |
Novavax |
Sanofi & GSK |
Vaccine Type |
mRNA |
mRNA |
Viral Vector |
Adenovirus |
Recombinant Protein Nanoparticle |
Adjuvanted Recombinant Protein |
Age Indication |
≥ 6 months old |
≥ 6 months old |
≥ 18 years old |
≥ 18 years old |
≥ 18 years old |
≥ 18 years old |
Vaccination Series |
Two vaccinations series and one booster A bivalent booster is indicated for patients > 5 |
Two vaccinations series and one booster A bivalent booster is indicated for patients > 6 |
One Vaccination and One Booster |
Two Vaccinations |
Two Vaccinations |
Still being assessed, likely two Vaccinations |
Use in Pregnancy & Lactation |
Yes |
Yes |
Yes |
Yes |
Use clinical judgment |
Unknown or insufficient available data |
Special Considerations |
Data not available |
Thrombosis-thrombocytopenia syndrome Guillain-Barre syndrome The FDA limited the authorized use of the J&J COVID-19 Vaccine to when mRNA vaccines are not accessible, clinically appropriate or patients will only receive the J&J vaccine. |
Data not available |
Data not available |
General Information on COVID-19 Vaccines
Vaccination During Pregnancy and Lactation
The World Health Organization’s Global Advisory Committee on Vaccine Safety found the benefits of mRNA COVID-19 vaccines outweigh the risks in reducing hospitalizations and deaths from COVID-19. The risk for myocarditis and pericarditis is very low. The estimate in patients 12 to 29 is 40.6 cases per million second doses among males and 4.2 cases per million among females. In patients over 30, the rate drops to 2.4 per million second doses in males and one per million second doses in females in patients. A preliminary FDA review found reported cases of myocarditis/pericarditis have been consistent with the Pfizer-BioNTech COVID-19 vaccine clinical trials. The reported cases were greater than expected in patients 16 to 24. After the second dose, the incidence is estimated to be 16 cases per million doses. A CDC analysis of EHR data from 40 health systems found the risk for myocarditis, pericarditis, or multisystem inflammatory syndrome to be 2 to 6 times higher in 12 to 17 year old boys who experienced a COVID-19 infection compared to receiving an mRNA COVID-19 vaccine. In young men 18 to 29 years, the risk is 7 to 8 times higher with infection over vaccination. In a review of VAERS data, the CDC found no increase in the occurrence of myocarditis with the Pfizer-BioNTech COVID-19 vaccine in males 5 to 11.
Omicron Variant and Vaccine Booster Doses
Pediatric COVID-19 Vaccines
FDA reviews of the Moderna and Pfizer- BioNTech mRNA COVID-19 vaccines found them to be as effective for children 6 months to 5 years as in older children and young adults. No cases of myocarditis were experienced in this age group during trials.
● Clearly labeling each prepared vaccine to differentiate adult and pediatric doses.
● Verifying each patient with two identifiers, such as name and age.
● Only bring one vaccine into the administration area at a time
● Document the lot and manufacturing date prior to administration and administration of the vaccine after it is given.
● Report all vaccine errors in VAERS. ISMP also request errors be reported to the ISMP VERP system
Moderna’s Vaccine is an mRNA vaccine with FDA approval for patients’ age 6-months old and up.
- The CDC’s ACIP recommended that COVID-19 vaccines from Pfizer and Moderna should be preferred over the J&J vaccine due to the risk for a rare but potentially fatal thrombosis with thrombocytopenia syndrome with the J&J vaccine.
- ACIP recommended that COVID-19 vaccines can be coadministered with other vaccines, including influenza vaccines in their 2021–22 Influenza Season update. A 679 patient, British Phase IV trial found that a COVID-19 vaccine given with an influenza vaccine on the same day in different arms will not affect the immune response of either vaccine. Adverse events were similar in patients that received both vaccines on the same day and patients that received one of the vaccines three-weeks later. The Pfizer-BioNTech and AstraZeneca COVID-19 vaccines were used in the study. Sanofi announced interim results from a trial where Sanofi’s Fluzone High-Dose Quadrivalent vaccine and Moderna’s COVID-19 mRNA investigational booster were co-administered. Immunogenicity and safety for both vaccines were similar to what is seen with either vaccine administered alone.
- ACIP met to review the safety of the three COVID-19 vaccines that have received an EUA. The group recommended that the benefits of vaccination outweigh the risks for Guillain-Barré syndrome and thrombosis with thrombocytopenia syndrome after Janssen COVID-19 vaccination and myocarditis after mRNA (Pfizer-BioNTech and Moderna) COVID-19 vaccination.
- The FDA may not review additional COVID-19 vaccines for emergency use if the sponsoring company is not already in discussions with the FDA. Novavax, Medicago and Astra Zeneca have discussed approval with the FDA. Other vaccine developers would need to seek full authorization.
- Researchers from Yale and the CDC analyzed COVID-19 vaccine efficacy in 463 nursing home residents during outbreaks at two Connecticut skilled nursing facilities. Vaccine efficacy was measured from 14 days after the first dose through seven days after the second, because there was not enough follow-up data to determine the impact of full vaccination. The researchers found the vaccines to be as effective as in the general adult population.
- A retrospective analysis of 513,284 patients, found the incidence of cerebral venous thrombosis to be eight times more common in patients that developed COVID-19 than in patients that received the AstraZeneca vaccine and ten times more common than in patients that received the Pfizer-BioNTech or Moderna vaccine.
- The CDC analyzed the safety data of the Pfizer-BioNTech or Moderna vaccines after 13,794,904 vaccine doses were given. The incidence of adverse events was 0.05% with the most common being headache, fatigue, and dizziness. Only 9.2% of adverse events were considered serious. The overall rate of anaphylaxis was similar to other vaccines with 4.5 events per million doses.
- In its COVID-19 vaccination recommendations, the CDC includes patients that have received the complete vaccination series with the AstraZeneca vaccine or the Novavax vaccine as being fully vaccinated and they do not also need immunization with a vaccine that is approved or available under an EUA
- British researchers analyzed the national British health database and found a higher incidence and prolonged duration for thrombocytopenia, venous thromboembolism and arterial thromboembolism leading to hospital admission or death in more people infected with COVID-19 than who received at least one vaccination with either the AstraZeneca or Pfizer-BioNTech vaccines.
- In the 1,072 patient, Phase III, Com-COV2 trial, patients received their first COVID-19 immunization with either the AstraZeneca or Pfizer-BioNTech vaccines. The second dose was given with the same vaccine, a vaccine from Moderna or Novovax. Both the Moderna and Novovax vaccines elicited higher antibody titers following the AZ vaccine than AZ’s own vaccine. Compared to a second dose of the Pfizer-BioNTech vaccine, Moderna elicited higher antibody titers, but Novovax did not.
- A systematic review and meta-analysis of 29 studies involving 11,713 solid organ transplant patients identified risk factors for decreased antibody titers. The risk factors included older age, recent transplantation, deceased donor status, active use of antimetabolites, and recent exposure to antithymocyte globulin or rituximab. Receiving additional doses of an mRNA vaccine increased the chance of developing higher antibody titers.
- Johns Hopkins’ researchers prospectively followed rheumatic and musculoskeletal diseases and solid organ transplant recipients patients who had been vaccinated with either the Pfizer-BioNTech or Moderna COVDI-19 vaccines. While the immune response was similar in patients not receiving immunosuppression the response was higher with the Moderna vaccine in patients receiving immunosuppressive drugs.
- A whole virus vaccine uses a weakened or inactivated virus
- Viral vectors use a genetically modified virus to produce a virus protein that causes the immune response. The immune response may be weakened if a common virus is used which has a high degree of immunity in the community. As an example, it is speculated that if a modified adenovirus is used, a booster immunization may be required to maintain immunity.
- Nucleic acid vaccines use DNA or RNA to causes a cell to produce a protein that causes an immune response. Since the full virus is not produced, no infection develops. No approved vaccine currently uses this technology. While mRNA and DNA vaccines can be designed, manufactured and tested faster than typical vaccines, the vaccine design have not been proven to be efficacious or safe.
- Protein-based vaccines use a protein from the virus that will cause an immune response without infection. This type of vaccine requires an adjuvant to stimulate the immune response. No protein-based vaccine has been tested in a human.
- Virus-like particles are empty virus shells that mimic the virus structure, but do not have the genetic material to cause an infection. These vaccines can stimulate a strong immune response but are harder to manufacture.
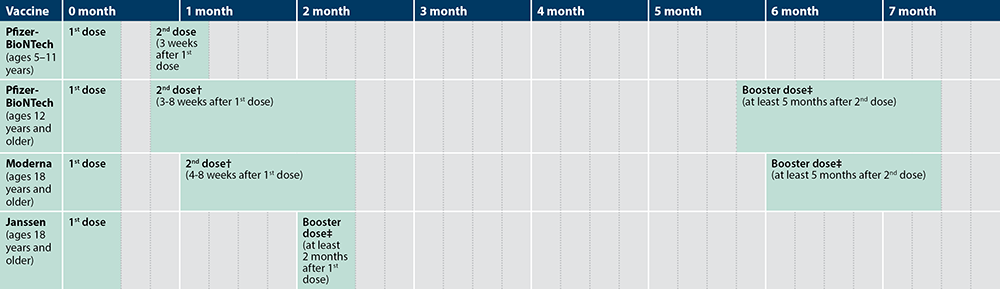
- Two doses of the Pfizer-BioNTech vaccine are given 21-days apart to patients 16 and older
- Two doses of the Moderna vaccine are given 28-days apart to patients 18 and older
- A single dose of the Johnson & Johnson vaccine is given 28-days to patients 18 and older
- Delay vaccination of patients with an acute COVID-9 infection until the patient has recovered. Immunization may be delayed for up to 90 days.
- Patients that have received convalescent or COVID-19 antibodies should have their immunization delayed for 90 days.
- Immunocompromised, autoimmune or pregnant patients with no contraindications, may receive the vaccine. Potential risks should be discussed with the patient, such as the unknown vaccine safety profile and effectiveness in immunocompromised populations, as well as the potential for reduced immune responses.
- Patients with a history of anaphylactic reactions should be observed for 30 minutes after vaccination
- Patients with a history of allergic, but not anaphylactic reactions should be observed for 15 minutes after vaccination
- No other vaccines should be given within two weeks of the Pfizer-BioNTech vaccine.
- Patients with a history of Guillain-Barré syndrome or Bell’s palsy may receive the vaccine.
- The agency continues to recommend the interval between doses, three weeks for the Pfizer-BioNTech vaccine and four weeks for Moderna’s vaccine, should be followed as closely as possible.
- If a patient is late for the second dose, they should receive the second vaccine by week six. There is not enough information to make a recommendation beyond six weeks, but an extra dose should not be administered if the second dose is not given by then.
- The Pfizer-BioNTech and Moderna vaccines are not considered interchangeable. However, if the initial vaccine a patient received cannot be determined or is not available, another mRNA vaccine may be given at least 28 days after the first vaccination.
- It is preferable for a patient that received an mRNA vaccine to receive a second dose with an mRNA vaccine. But if no mRNA vaccine is available or there is a contraindication to receiving one, then the patient can receive the Johnson & Johnson vaccine for the second dose at least 28-days after the initial vaccination.
Vaccination During Pregnancy and Lactation
- The American College of Obstetricians and Gynecologists (ACOG) recommends the Pfizer-BioNTech, Moderna or Johnson & Johnson COVID-19 vaccines should not be withheld from pregnant or lactating women who meet criteria for vaccination based on ACIP-recommended priority groups. A discussion with the patient of the vaccine should include the potential for adverse effects, hand washing and wearing a mask. Patients should be educated that an mRNA vaccine is not a live virus vaccine and no adjuvant is used in the Pfizer-BioNTech COVID-19 vaccine. The mRNA vaccines do not enter the nucleus and do not alter human DNA in vaccine recipients, so these types of vaccines cannot cause any genetic changes. The CDC also discusses mRNA vaccines on a dedicated web page. ACOG recommends the patient’s decision on whether to receive the vaccine should be supported.
- The CDC analyzed surveillance data from 30,494 patients who were vaccinated with either the Pfizer-BioNTech or Moderna COVID-19 vaccines during pregnancy and found the rates of complications were not significantly different from those of unvaccinated pregnant women.
- Researchers examined data from 30 pregnant, 16 lactating, and 57 non-pregnant/non-lactating patients and found the mRNA vaccine response was equivalent for pregnant and lactating women compared to non-pregnant/non-lactating women. Further, the immune transfer to neonates occurred via placental and breastmilk. Pregnant and lactating women also developed neutralizing antibodies that were active against the B.1.1.7 (U.K.) and B.1.351 (South African) variants.
- Researchers analyzed data from 35,691 pregnant women, who had received either the Pfizer–BioNTech or Moderna COVID-19 vaccines, that were enrolled in the V-safe Surveillance System and Pregnancy Registry and the Vaccine Adverse Event Reporting System (VAERS). Compared to nonpregnant women the incidence of injection-site pain was more frequent, while headache, myalgia, chills, and fever were less frequent. The rate of spontaneous abortion was 12.6%, which falls within the historical range of 10% to 26%. While not directly comparable, the rate of adverse pregnancy and neonatal outcomes after COVID-19 vaccination were similar to the rates reported before the COVID-19 pandemic. Based on data from the study, the CDC now recommends COVID-19 vaccination for pregnant women.
- A retrospective analysis of the outcomes of 15,060 pregnant women in an Israeli database found the estimated effectiveness of the Pfizer-BioNTech COVID-19 vaccine to be 78%.
- A study of 36 women, who received an mRNA vaccine during pregnancy, found that antibodies were passed on to infants.
- A case-control study involving 105,446 pregnancies, from eight large U.S. healthcare systems and an analysis of outcomes from 2,456 women enrolled in a CDC pregnancy registry found no increase in the risk for spontaneous abortion with COVID-19 vaccinations.
- An analysis of the response to vaccination with mRNA vaccines found that pregnant and lactating women develop lower antibody titers after the first vaccine dose. Antibody levels rise to match the immunological response seen in non-pregnant and non-lactating women after the second dose.
- A Norwegian case–control study examined first-trimester pregnancies in 13,956 women from registry data and found no evidence of an increased risk for early pregnancy loss after Covid-19 vaccination.
- A retrospective analysis of maternal and umbilical cord blood samples from 1,359 vaccinated pregnant women found that COVID-19 immunization before and throughout pregnancy resulted in detectable antibodies at delivery. The highest maternal and umbilical cord antibody levels were found in mothers who had received a complete vaccination course, had a prior history of COVID-19, or received a third-trimester booster dose.
- An analysis of COVID-19 antibodies in breast milk of vaccinated mothers found that 96% of lactating women who received the Pfizer-BioNTech vaccine and 97% of mothers that received the Moderna vaccine had detectable IgA antibodies in their milk compared to 39% who received the AstraZeneca vaccine and 48% who received the Johnson & Johnson vaccine.
- A Canadian retrospective analysis of 97,590 pregnant patients and a retrospective study of 157,521 singleton births in Sweden and Norway found that COVID-19 vaccination during pregnancy did not increase the risk of adverse peripartum outcomes compared with vaccination after pregnancy and with no vaccination. Most patients received an mRNA vaccine during the second and third trimester in both studies.
- A pre-print draft of an Israeli analysis examining SARS-CoV-2 antibodies during pregnancy, found antibodies levels were too low to provide protection by delivery in 34.6% of patients who experienced a first trimester infection and 9.1% who experienced a second trimester infection. A single dose of the Pfizer COVID-19 vaccine induced protective antibody titers for both mother and infant with modest adverse effects.
- A test-negative, case-control study by the CDC and academic partners, of 5,492 health care encounters for pregnant people, found the vaccine effectiveness (VE) for two doses to prevent COVID-19–associated emergency department and urgent care encounters during Omicron predominance was not significant, but VE for three doses was 79%. VE to prevent COVID-19 associated hospitalizations with was 86% for up to five months with two doses and up to four months with three doses.
The World Health Organization’s Global Advisory Committee on Vaccine Safety found the benefits of mRNA COVID-19 vaccines outweigh the risks in reducing hospitalizations and deaths from COVID-19. The risk for myocarditis and pericarditis is very low. The estimate in patients 12 to 29 is 40.6 cases per million second doses among males and 4.2 cases per million among females. In patients over 30, the rate drops to 2.4 per million second doses in males and one per million second doses in females in patients. A preliminary FDA review found reported cases of myocarditis/pericarditis have been consistent with the Pfizer-BioNTech COVID-19 vaccine clinical trials. The reported cases were greater than expected in patients 16 to 24. After the second dose, the incidence is estimated to be 16 cases per million doses. A CDC analysis of EHR data from 40 health systems found the risk for myocarditis, pericarditis, or multisystem inflammatory syndrome to be 2 to 6 times higher in 12 to 17 year old boys who experienced a COVID-19 infection compared to receiving an mRNA COVID-19 vaccine. In young men 18 to 29 years, the risk is 7 to 8 times higher with infection over vaccination. In a review of VAERS data, the CDC found no increase in the occurrence of myocarditis with the Pfizer-BioNTech COVID-19 vaccine in males 5 to 11.
Omicron Variant and Vaccine Booster Doses
- On 3/29/2022, the FDA approved a fourth dose (second booster) of mRNA #COVID-19 vaccines for patients 50 and older. In certain immunocompromised patients the age is lowered to12 and older with the Pfizer-BioNTech vaccine and 18 and older with the Moderna vaccine. The CDC endorsed the new approval and recommended certain immunocompromised individuals and people over the age of 50 who received an initial booster dose at least 4 months be eligible for a second booster (fourth dose) or an mRNA COVID-19 vaccine. These decisions were made before a 4/6/2022 meeting to discuss vaccine boosters. The FDA's Vaccines and Related Biological Products Advisory Committee (VRBPAC) did not develop a plan for COVID-19 vaccine booster composition or timing of additional boosters. In addition to attempting to predict which variant will be dominant, half of the U.S. population has not received a booster dose, so how to optimize protection for everyone will be more of a challenge. VRBPAC members agreed the goal for booster doses should be prevention of hospitalization and death in at least 80% of patients. Peter Marks, the director of the FDA’s Center for Biologics Evaluation and Research, said the fourth dose vaccine dose was a reasonable approval until a new booster, that would preferably have longer-lasting protection, was available. He felt frequent use of boosters was not a strategy that should be continued. VRBPAC will meet again to discuss more specific details of a booster program for COVID-19.
- An Israeli study involving 1,050 healthcare workers, who had received a Pfizer/BioNTech or Moderna COVID-19 booster dose at least four months earlier, found an increase in the immune response with a fourth dose against the omicron variant. However, vaccine efficacy was only 30% for any severity of COVID-19 with most cases being mild.
- An analysis of EHR data from 1,252,331 Israeli patients, who were 60 and older, found that a fourth vaccination with an mRNA COVID-19 vaccine lowers the risk by half for a confirmed infection. But the protection wanes and only lasts about eight weeks. Protection against severe cases of COVID-19 were three times lower and did not appear to decrease over time. The study was too short to estimate the duration of protection against severe disease.
- A CDC analysis found that mRNA COVID-19 vaccine effectiveness for the Omicron variant, two months after a booster dose, was 87% to prevent emergency room/urgent care visits (ED/UC) and 91% to prevent hospitalizations. Four months after the booster dose vaccine, effectiveness was 66% to prevent ED/UC and 91% to prevent hospitalizations. A separate study found that mRNA vaccines are 94% effective, after three doses, to prevent invasive mechanical ventilation from severe COVID-19 caused by the Omicron variant.
- An academic analysis of mRNA COVID-19 vaccine effectiveness to prevent hospitalizations, for several SARS-CoV-2 variants, found vaccine effectiveness to be 65% after two immunizations and 85% after three for Omicron. Severity was lower when comparing Delta to Omicron infections. Patients who were vaccinated also had less severe disease for all variants.
- A CDC analysis of data from the Increasing Community Access to Testing (ICATT) national SARS-CoV-2 testing program, from September 14–November 11, 2022, found the Moderna and Pfizer/BioNTech bivalent vaccines to be more effective than the original vaccines. Vaccine effectiveness for the original vaccine compared to the bivalent vaccine were estimated to be: 30% compared to 56% for patients 18–49 years, 31% compared to 48% for patients 50–64 years and 28% compared to 43% for patients 65 years or older
- An FDA review of data available for the Omicron variant vaccines from Moderna and Pfizer-BioNtech found that both vaccines improved the neutralizing antibody response to Omicron BA.1. The FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) voted 19-2, on 6/28/2022, to recommend incorporating a component of the omicron variant into future COVID booster vaccines. The Omicron BA.4 and BA.5 subvariants now comprise more than 50% of COVID-19 cases. If the FDA requests a new booster vaccine to include these variants, Moderna and Pfizer-BioNTech would be able to create the vaccines by October. VRBPAC recommended a new booster be bivalent and contain BA.4 or BA.5 subvariants of Omicron. The FDA amended the EUAs for the Moderna and Pfizer-BioNTech COVID-19 bivalent booster vaccines to include children and adolescents. Both vaccine can be given at least two months after the last booster vaccination. The Pfizer-BioNTech COVID-19 Vaccine is approved for patients five years and older. The Moderna vaccine is approved for patients six years and older.
- The FDA found that an investigational 50 mcg bivalent vaccine from Moderna, used as a fourth dose, elicited 75% higher antibodies for the Omicron BA.1 subvariant.
- An investigational 30 mcg Omicron specific vaccine from Pfizer-BioNTech elicited 56% more antibodies. Pfizer-BioNTech presented data that demonstrated activity against the BA.4 and BA.5 subvariants in mice.
- An investigational COVID-19 vaccine from Novavax, that uses a protein-based vaccine, demonstrated antibodies that remained active against Omicron and its subvariants. Novavax will not have data on an Omicron specific vaccine until September.
- Moderna announced additional interim data from an 800 patient, Phase II/III trial, where an investigational bivalent COVID-19 vaccine, containing a combination of the original vaccine and an Omicron variant specific vaccine, used as a booster dose, increased neutralizing antibodies against BA.4 and BA.5 by 5.4-fold in all participants regardless of prior infection.
- Pfizer and BioNTech announced that in a 1,234 patient, Phase II/III trial, an investigational Omicron specific vaccine, given as a fourth booster dose, increased neutralizing antibody titers by 13.5 with a 30 mcg dose and 19.6 fold with a 60 mcg dose against Omicron BA.1. A vaccine that combined the Omicron vaccine with the original vaccine increased neutralizing antibody titers by 9.1 and 10.9-fold increase against Omicron with 30 mcg and 60 mcg doses.
Pediatric COVID-19 Vaccines
FDA reviews of the Moderna and Pfizer- BioNTech mRNA COVID-19 vaccines found them to be as effective for children 6 months to 5 years as in older children and young adults. No cases of myocarditis were experienced in this age group during trials.
- The Pfizer- BioNTech review estimated a three-dose regimen of the Pfizer-BioNTech to be 75.6% effective in infants 6 to 23 months and 82.4% for toddlers 2 to 4 years during an Omicron variant dominant period.
- The Moderna review estimated the vaccine effectiveness for a two-dose regimen of the Moderna vaccine to be:
- •93.3% for adolescents 12 to 17 years
- •76.8% for children 6 to 11 years
- •36.8% for toddlers 2 to 5 years
- •50.6% for infants 6 to 23 months
- •Only the results for children 6 months to 5 years were during an Omicron dominant period. The results are based on preliminary results and may not represent the actual effectiveness.
- The FDA’s Vaccine and Related Biologics Advisory Committee voted 21 to 0 to recommend an EUA for the Pfizer-BioNTech COVID-19 vaccine for infants and toddlers 6 months to 4 years. The FDA granted an EUA for the Pfizer-BioNTech COVID-19 vaccine on 6/17/2022 for infants and toddlers 6 months to 4 years.
- The Advisory Committee also voted 21 to 0 to recommend an EUA the Moderna COVID-19 vaccine for children 6 months to 17 years. The FDA granted an EUA for the Moderna COVID-19 vaccine on 6/17/2022 for children and adolescents 6 months to 17 years.
- The CDC’s Advisory Committee on Immunization Practices’ (ACIP) added a recommendation to their COVID-19 immunization guidance for all children 6 months through 5 years of age. The CDC endorsed the recommendation on 6/18/2022.
- The FDA amended the EUAs for the Moderna and Pfizer-BioNTech COVID-19 bivalent booster vaccines to include children and adolescents. Both vaccine can be given at least two months after the last booster vaccination. The Pfizer-BioNTech COVID-19 Vaccine is approved for patients five years and older. The Moderna vaccine is approved for patients six years and older.
- Two doses of BNT162 are given 21 days apart with a booster given five months after the second dose.
- Immunocompromised patients should receive a total of 4 doses of an mRNA COVID-19 vaccine. Currently the CDC recommends that those who are ages 12 years and older and are moderately or severely immunocompromised should receive a primary series of 3 doses of an mRNA COVID-19 vaccine plus 1 mRNA booster. A series of 3 doses is recommended for immunocompromised patients under the age of 12, but no recommendation for a booster at this time.
- While children seem to be less affected by COVID-19 than adults, there has been 8,300 hospitalizations for COVID-19 in children ages 5 to 11 and 146 children have died. In the same age group, 2,316 children have developed multisystem inflammatory syndrome (MIS-C) from COVID-19 and nine have died. The mortality rate with COVID-19 in pediatric patients is much higher than influenza. The CDC noted a five-fold increase in pediatric COVID-19 cases attributed the Delta variant in summer 2021. CDC researchers reviewed data from VAERS and V-Safe voluntarily surveillance systems for children ages 5 to 11 years from 11/3/2021 to 12/19/2021. The researchers found the most common adverse reactions were injection site pain, fatigue and headache. ADR were mostly mild, and few cases of myocarditis were reported. It was estimated that 8.7 million doses were given during this time period to children 5 to 11 years.
- Pfizer and BioNTech announced that in 4,500 patient, Phase I/II/III trial (NCT04816643), two 3 mcg doses of their COVID-19 vaccine given to patients 6 to 24 months old demonstrated immunogenicity similar to that seen with the adult dose in patients 16 to 25 years old. An inadequate response was elicited in patients 2 to under 5 years old with the 3 mcg dose, so a third dose will be evaluated in patients 6 months to under 5 years. Pfizer-BioNTech announced that in a 1,678 patient, Phase II/III trial, vaccine efficacy after three 3mcg doses of their COVID-19 vaccine in children six months to five years was 80.3%. Antibody titers were similar in young children as in patients 16 to 25 years. The third dose was given two months after the second and the trial was done when Omicron was the dominant variant.
- A CDC test-negative, case-control study, using data from December 2021 to February 2022, estimated the Pfizer-BioNTech COVID-19 vaccine effectiveness against symptomatic infection for children 5 to 11 years as 60% one month after the second vaccine dose and 29% after two months. Vaccine effectiveness for adolescents 12 to 15 years of age was 60% one month after the second dose and 17% after two months. Vaccine effectiveness for adolescents who received a booster dose was 71%.
- The CDC analyzed the safety data of the Pfizer-BioNTech or Moderna vaccines after 13,794,904 vaccine doses were given. The incidence of adverse events was 0.05% with the most common being headache, fatigue, and dizziness. Only 9.2% of adverse events were considered serious. The overall rate of anaphylaxis was similar to other vaccines with 4.5 events per million doses.
- The FDA has advised it is acceptable to use each additional full dose in the vials. But partial doses from different vials, should not be combined for a full dose, since the vials are preservative free.
- Researchers estimate the incidence of anaphylaxis with the Pfizer-BioNTech COVID-19 vaccine as 11.1 cases per million doses administered.
- An analysis of patients with immune-mediated inflammatory diseases who were receiving immunomodulatory treatments found that use of methotrexate decreased the immune response from the Pfizer-BioNTech COVID-19 vaccine. Over 90% of patients that were receiving a TNF blocker achieved a robust antibody response compared to only 62.2% of patients that were receiving methotrexate that achieved an adequate response.
- In a review of data from 1,137,804 patients from the Israeli Ministry of Health database, found that a booster dose of the Pfizer/BioNTech COVID-19 vaccine, given at least five months after the second dose in patients 60 years or older, resulted in a reduction in confirmed and severe cases of COVID-19, during the first 12 days after the third dose.
- A third (booster) dose of the Pfizer/BioNTech COVID-19 vaccine was given to the pivotal trial population (NCT04368728), 7.9 to 8.8 months after the second dose. Antibody titers increased after the third dose for the original SARS-CoV-2 strain and variants.
- South African researchers examined the results of 133,437 COVID-19 PCR tests and found the effectiveness of the Pfizer-BioNTech COVID-19 vaccine to be 70% after two doses in preventing hospitalizations for COVID-19 during a period when Omicron variant was the dominant strain compared to 93% when the Delta variant was the dominant strain.
- In a second study, South African researchers compared the COVID-19 neutralization ability for the Beta, Delta, and Omicron variants with serum from 20 patents that had received two vaccinations with the Pfizer-BioNTech COVID-19 vaccine to a group of 20 patients that had received three injections. The researchers found that a third dose of the vaccine increased neutralization efficiency by a factor of 100 against the Omicron variant, but neutralization was lower by a factor of four against the omicron variant compared to the delta variant.
- Pfizer and BioNTech announced immunology data from a 140 patient, Phase II/III trial, where a booster dose of their COVID-19 vaccine, given six-months after the initial series, produced a six-fold increase in antibodies one month after the booster compared to antibody titers one month after the second dose in children 5 through 11 years of age. A sub-analysis of 30 patients found that the Omicron variant was neutralized by the antibodies.
- Pfizer and BioNTech announced early interim data from a 900 patient, Phase II/III trial (NCT05472038), which demonstrated an increase in antibody titers for the Omicron BA.4 and BA.5-subvariants seven-days after immunization with their bivalent booster vaccine.
- Pfizer and BioNTech announced early interim data from a 900 patient, Phase II/III trial (NCT05472038), which demonstrated an increase in antibody titers, one-month after immunization with their bivalent booster vaccine, for the Omicron BA.4 and BA.5-subvariants by 13.2-fold in adults over 55 years and 9.5-fold in adults 18 to 55 years of age, compared to a 2.9-fold increase in adults over 55 who received the original booster vaccine.
- A pre-print draft described how one month after a booster dose with the new bivalent Pfizer/BioNTech COVID-19 vaccine, antibody titers were increased for Omicron sublineages BA.4/5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 compared to a booster dose with the original vaccine. The increase in antibodies with the bivalent vaccine was true in patients whether or not they had a past COVID-19 infection.
- ISMP issued a warning regarding use of an incorrect dose of the Pfizer-BioNTech COVID-19 vaccine. The pediatric vaccine (10 mcg/0.2 ml) has been used in patients 12 and older and the adult dosage (30 mcg/0.3 ml) has been used in patients under 12. ISMP further warns against diluting the adult vaccine to make a pediatric vaccine. Due to the vaccine being a suspension and the small volume, it would not be possible to create an accurate dose. To avoid errors, ISMP recommends
● Clearly labeling each prepared vaccine to differentiate adult and pediatric doses.
● Verifying each patient with two identifiers, such as name and age.
● Only bring one vaccine into the administration area at a time
● Document the lot and manufacturing date prior to administration and administration of the vaccine after it is given.
● Report all vaccine errors in VAERS. ISMP also request errors be reported to the ISMP VERP system
Moderna’s Vaccine is an mRNA vaccine with FDA approval for patients’ age 6-months old and up.
- The vaccine is given as two Intramuscular Injection given 28 days apart.
- Immunocompromised patients should receive a total of 4 doses of an mRNA COVID-19 vaccine. Currently the CDC recommends that those who are ages 12 years and older and are moderately or severely immunocompromised should receive a primary series of 3 doses of an mRNA COVID-19 vaccine plus 1 mRNA booster. A series of 3 doses is recommended for immunocompromised patients under the age of 12, but no recommendation for a booster at this time.
- The CDC estimates the incidence of anaphylaxis with Moderna’s vaccine to be 2.5 cases per million doses administered.
- Physicians at Massachusetts General Hospital describe delayed cutaneous reactions in 12 patients, who were given the Moderna COVID-19 vaccine. The reactions happened around eight days after the initial immunization and lasted about six days. All patients received their second dose of vaccine and six of the patients developed a second reaction within two days. While the cutaneous reaction is rare, it is more likely to be seen during a mass vaccination campaign, due to the large number of vaccinations. A retrospective study by Yale identified 16 patients that developed a delayed localized cutaneous reactions 2 to 12 days after receiving the Moderna COVID-19 vaccine. The reaction was near the injection site and was pruritic and painful with edematous pink plaques. The reaction developed in 11 of the patients with the second dose. The reaction was also seen in a small number of patients during the pivotal COVE trial. The reaction had a duration of five days.
- In a four-month, 120 patient, Phase IV trial (NCT04885907), 55% of patients that received a third dose of the Moderna COVID-19 vaccine achieved at least 100 U/ml of anti–receptor-binding domain (RBD) compared to 18% that received only two doses in solid organ transplant patients that did not have a previous diagnosis of COVID-19. The percent virus neutralization was 71% in the three-dose group compared to 13% in the two-dose group.
- Moderna announced interim data from a cohort of 4,753 children, age 6 to under 12 years of age, enrolled in the Phase II/III KidCOVE trial (NCT04796896), where two 50 mcg doses of Moderna’s COVID-19 vaccine, given 28-days apart, elicited a strong immune response and was well tolerated.
- In the 4,016 patient, Phase II/III KidCOVE trial (NCT04796896), where two 50 mcg doses of Moderna’s COVID-19 vaccine, given 28-days apart, elicited immunogenicity in children 6 to under 12 years of age that was similar to young adults, age18 to 25 years, who received two 10 mcg doses. Vaccine efficacy was estimated to be 88% during a period when the delta variant was dominant.
- Moderna announced that preliminary lab results found that antibody levels after a booster (third) dose of their COVID-19 vaccine increased antibody levels that are thought to be high enough to neutralize the Omicron variant. A pre-print draft described how antibody levels were not high enough after two doses of the vaccine to neutralize the Omicron variant.
- A pre-print draft describes an 895 patient, open-label, Phase II/III trial, where a bivalent COVID-19 vaccine developed by Moderna was tested against its approved vaccine. The bivalent vaccine consisted of the original vaccine with the addition of mRNA for the Beta variant spike proteins. Compared to original vaccine, the bivalent vaccine elicited increased antibody titers for the original virus and Beta, Omicron and Delta variants after 28 days. Antibody titers levels were still elevated for the bivalent vaccine at 180 days for the original Beta, Omicron variants. The bivalent vaccine was given as a booster about nine months after the primary series. Moderna is also developing a bivalent vaccine that combines the original vaccine with mRNA for the Omicron variant spike proteins. A blinded randomized trial for this vaccine will be reported in 2Q22. Moderna feels the Omicron bivalent vaccine will be chosen for use as a booster in the fall.
- In a 437 patient, Phase II/III trial (NCT04927065), Moderna’s bivalent COVID-19 vaccine, increased neutralizing antibodies against all Omicron subvariants, including BA.4 and BA.5 compared to the original Moderna vaccine in all participants regardless of prior infection.
- Moderna announced an increase in antibody titers with its bivalent booster vaccine for the BQ.1.1 subvariant, though the increase was 5-fold less than for the Omicron BA.4/BA.5 subvariants.
- The FDA limited the authorized use of the J&J COVID-19 Vaccine to when mRNA vaccines are not accessible or clinically appropriate or for patients who choose to receive the vaccine and would otherwise not receive a COVID-19 vaccination. The FDA considers the known and potential benefits of the vaccine to outweigh the known and potential risks of receiving it. The restriction was due to the rare occurrence of thrombosis with thrombocytopenia syndrome (TTS). TTS is estimated to have an incidence of 3.23 cases per million vaccinations and 0.48 deaths per million. Risk factors for TTS associated with the J&J vaccine have not been identified and prompt diagnosis and treatment of TTS may not prevent deterioration of the condition.
- Patients who are moderately or severely immunocompromised and have received a Johnson and Johnson COVID-19 vaccine are recommended to receive a second dose of either Pfizer-BioNTech or Moderna’s mRNA COVID-19 vaccine, as well as, a booster for a total of 3 doses.
- The EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) recommended adding a warning regarding a rare adverse event of thrombosis in combination with thrombocytopenia to the product label for the Johnson & Johnson COVID-19 vaccine. PRAC reviewed eight cases of thrombosis with thrombocytopenia among seven million patients that received the vaccine in the U.S. The committee noted that all cases happened within 3 weeks of receiving the vaccine in patients under 60. Most cases occurred in women and were very similar to those seen with the AstraZeneca vaccine.
- Researchers analyzed historical and current data from Olmsted County, Minnesota and found the incidence of cerebral venous sinus thrombosis (CVST) had increased in females who received the J&J COVID-19 vaccine. The highest incidence of CVST was in women 40-49, followed by women aged 30 to 39 years.
- NVX-CoV2373 is also listed as SARS-CoV-2 rS.
- The FDA issued an EUA for the Novavax COVID-19 Vaccine, on 7/13/2022, to prevent COVID-19 in patients 18 years of age and older. The Novavax COVID-19 Vaccine is administered as a two-dose primary series given three weeks apart.
- The FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) voted 21-0 with one abstention to recommend an EUA for Novavax’s two-dose COVID-19 vaccine for patients 18 years and older. An FDA review and the discussion by the VRBPAC expressed concern about myocarditis, although the adverse effect appears to be rare.
- In a 14,039 patient, Phase III trial (EudraCT number, 2020-004123-16), two doses of NVX-CoV2373 given 21-days apart resulted in 89.7% efficacy based on 106 cases of moderate to severe COVID-19 that developed in the United Kingdom study population. Five cases of severe disease occurred, and all were in the placebo group. Efficacy was 96.4% against the original COVID-19 strain and 86.3% against the B.1.1.7 (UK) variant.
- In a 4,387 patient, Phase IIb trial (NCT04533399), where two doses of the Novavax COVID-19 vaccine given 21-days apart resulted in 49.4% efficacy in a South African population. Most cases of COVID-19 were the B.1.351 (South African) variant. The trial population included medically stable, HIV-positive adults. Efficacy was 60.1% among HIV negative patients.
- In the 29,582 patient, Phase III, PREVENT-19 trial (NCT04611802), two immunizations of NVX-CoV2373 (Novavax)ngiven 21-days apart resulted in 90.4% efficacy for developing a COVID-19 infection and 100% efficacy in preventing severe COVID-19.
- Novavax has added crossover arms to its studies to allow trial participants that received placebo to get the vaccine without unblinding the trials. A second round of vaccines will be offered for participants in the 15,000 patient, U.K. trial (NCT04583995) and the 30,000 patient, Phase III, PREVENT-19 trial (NCT04611802) in the U.S. and Mexico. Patients that received the vaccine will receive placebo and placebo patients will receive the vaccine. In the 2,905 patient, South African trial (NCT04533399), placebo patients will receive the vaccine, while the original vaccine patients will receive a booster to determine if a booster will improve efficacy for the B.1.351 (South African) variant.
- Novavax announced preliminary data demonstrating that a single booster dose of NVX-CoV2373, given six months after the initial two-dose regimen resulted in a 4.6-fold increase in antibody titers.
- Novavax announced that in a 14-day, 2,090 patient, Phase III trial (NCT05372588), a bivalent booster targeting the BA.1 and BA.5 omicron strains did not elicit more antibodies than Novavax’s current vaccine or a monovalent vaccine aimed solely at BA.1.
- AZD1222 is also listed as ChAdOx1 nCoV-19
- The World Health Organization (WHO) has endorsed the AstraZeneca COVID-19 vaccine and recommends two doses, given eight to 12 weeks apart.
- AstraZeneca is designing a new trial to evaluate the efficacy of the half-strength initial dose/full strength second dose regimen that demonstrated 90% efficacy in a small subpopulation in a 22,690, Phase III trial.
- Some countries have expressed concern for an increased risk for thromboembolic events with the AstraZeneca COVID-19 vaccine. The EMA’s Pharmacovigilance Risk Assessment Committee (PRAC) found the AstraZeneca COVID-19 vaccine causes a rare adverse event of thrombosis in combination with thrombocytopenia. PRAC reviewed 62 cases of cerebral venous sinus thrombosis and 24 cases of splanchnic vein thrombosis and concluded the thrombi are most likely to occur within the first two weeks after administration in women younger than 60. PRAC advises the benefits of the vaccine continue to outweigh the risks and the vaccine is effective at preventing COVID-19 and reducing hospitalizations and deaths. The U.K.’s Medicines and Healthcare products Regulatory Agency (MHRA) has recommended that while prompt vaccination with the AZ vaccine outweigh the risk of thrombosis, patients under 30 should be offered an alternative vaccine. The World Health Organization has stated that a causal relationship between the rare event of thrombosis in combination with thrombocytopenia had not been proven, but surveillance and investigation should be continued. German researchers found evidence the rare immune thrombotic thrombocytopenia is mediated by platelet-activated PF4 antibodies that is clinically similar to autoimmune heparin-induced thrombocytopenia. Norwegian researchers found similar evidence in a separate group of patients. British researchers analyzed the national British health database and found a higher incidence and prolonged duration for thrombocytopenia, venous thromboembolism and arterial thromboembolism leading to hospital admission or death in more people infected with COVID-19 than who received at least one vaccination with either the AstraZeneca or Pfizer-BioNTech vaccines.
- Sanofi and GSK announced a delay in their COVID-19 vaccine development program when an interim analysis of a 441 patient, Phase I/II trial (NCT04537208) found the vaccine to elicit antibodies comparable to convalescent plasma in patients 18 to 49, but the response in older patients was insufficient.
- Sanofi and GSK announced interim results from a 722 patient, Phase II trial (NCT04762680), where the companies’ COVID-19 vaccine elicited antibody levels comparable to recovered patients following a second dose in all age groups (18 to 95 years old) of patients from the U.S. and Honduras. Based on the interim results from the Phase II trial, Sanofi and GSK are planning to initiate a 35,000 patient Phase III trial with the goal of submitting data for an emergency use authorization (EUA) in 4Q21.
- Sanofi and GSK announced that in a 521 patient, Phase I/II trial (NCT04537208), using their COVID-19 vaccine as a booster dose for vaccines from Pfizer/BioNTech, Moderna, Johnson & Johnson, and AstraZeneca resulted in an increase in neutralizing antibodies of 9- to 43-fold.
- Sanofi and GSK announced that in the 1,500 patient, Phase III, VAT02 trial (NCT04762680), vaccination with their COVID-19 booster vaccine after an mRNA vaccine doubled antibody titers for Omicron BA.1 and BA.2 compared to the their original booster vaccine.
- Sanofi and GSK announced that in the 13,000 patient, Phase III, VAT08 trial (NCT04904549), primary vaccination with two doses of their bivalent COVID-19 vaccine resulted in vaccine efficacy of 64.7% against any symptomatic COVID-19 disease, 72% effectiveness in preventing Omicron variant symptomatic cases.
- Sanofi and GSK announced that in the 247 patient, Phase III, COVIBOOST trial (NCT05124171), a booster dose after two doses of the Pfizer-BioNTech COVID-19 vaccine resulted in a 10-fold increase in antibody titers in:
- 76.1% who received the Sanofi-GSK next-generation booster.
- 63.2% who received a booster with a third dose of the Pfizer-BioNTech vaccine.
- 55.3% who received the original Sanofi-GSK booster candidate.